fetchGrinnCorrNetwork
This example shows how to reconstruct a biological network (grinn network) using information from Grinn internal database, then compute and combine with a weighted correlation network
- INPUT
The table summarizes important arguments
Argument Value Description txtInput list of keywords e.g. ENSG00000143811 Keywords are IDs from a specific database. from type of keyword e.g. metabolite Type of starting points in the network. It can be one of metabolite, protein, gene, pathway. to type of end nodes e.g. pathway Type of endpoints in the network. It can be one of metabolite, protein, gene, pathway. datX data frame containing normalized-omic data Columns correspond to entities e.g. genes and rows to samples e.g. normals, tumors. Require 'nodetype' at the first row to indicate the type of entities in each column. datY data frame containing normalized-omic data It uses the same format as datX. Or it can be NULL, if there is only one omic dataset corrCoef numerical value from 0-1 The minimum absolute correlations to include edges in the output pval numerical value The maximum pvalues, to include edges in the output - EXECUTE FUNCTION
Build a grinn network of pathway-metabolite using the list of SMPDB pathways, then combine with a correlation network of metabolites
#1. load metabolomics data from a csv file datMet = read.csv("Lung_MET.csv", header=TRUE, row.names=1, stringsAsFactors=FALSE) #2. show dimensions of metabolomics data dim(datMet) #3. show the first 10 rows and 10 columns datMet[1:10,1:10] #!!--- NOTE: The following codes convert kegg ids to grinn ids. If the input data is already the grinn ids, these steps can be skipped. grinnID = convertToGrinnID(txtInput=colnames(datMet), nodetype="metabolite", dbXref="kegg") #call grinn function to convert ids grinnID = grinnID[!duplicated(grinnID[,1]),] #keep the first mapped id colnames(datMet) = lapply(colnames(datMet),function(x) ifelse(length(which(grinnID$FROM_kegg == x))>0,as.character(grinnID$GRINNID[which(grinnID$FROM_kegg == x)]),x)) #---------- END id conversion ----------# #4. load pathway keywords from a text file (.txt) ptwKw = unlist(read.csv("pathwayList.csv", stringsAsFactors=FALSE, header=FALSE)) #5. show length of keywords length(ptwKw) #6. show the first 2 keywords ptwKw[1:2] #7. execute function result <- fetchGrinnCorrNetwork(txtInput=ptwKw, from="pathway", to="metabolite", datX=datMet, corrCoef=0.7, pval=1e-5, method="spearman", dbXref = "smpdb") #display the first 10 edgelists result$edges[1:10,] - EXPORT OUTPUT
Export the network as tab-delimited files to visualize in Cytoscape
write.table(as.matrix(result$edges),"grCorrNwEdge.txt",sep="\t",row.names = F, quote = FALSE) write.table(as.matrix(result$nodes),"grCorrNwNode.txt",sep="\t",row.names = F, quote = FALSE) - VISUALIZATION
The figure is generated by Cytoscape 3.1.1 using grinn style (grinn.xml). It is corresponding to the cytoscape file grCorrNw.cys.
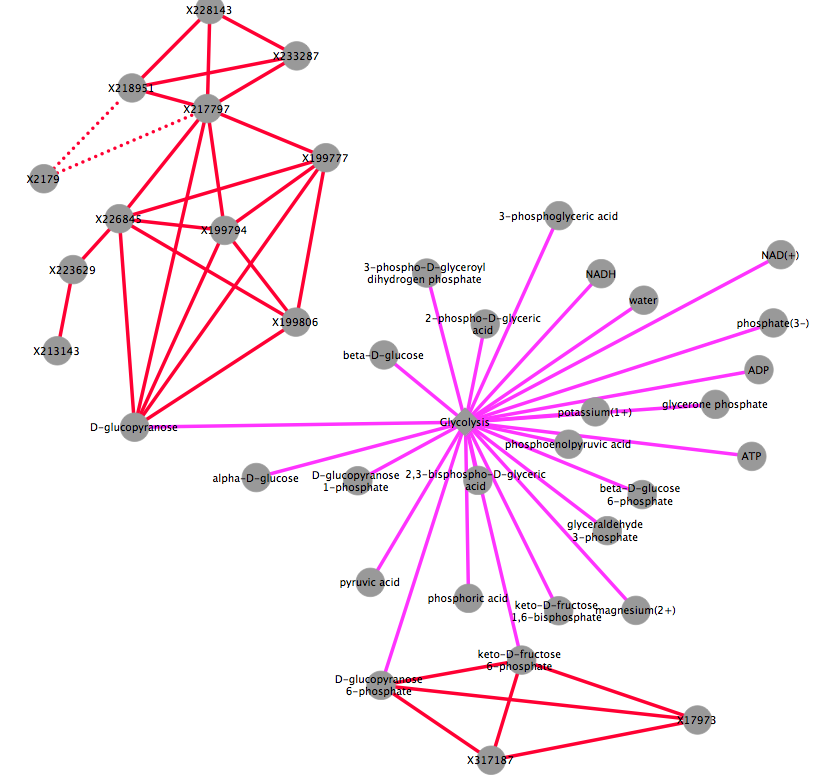
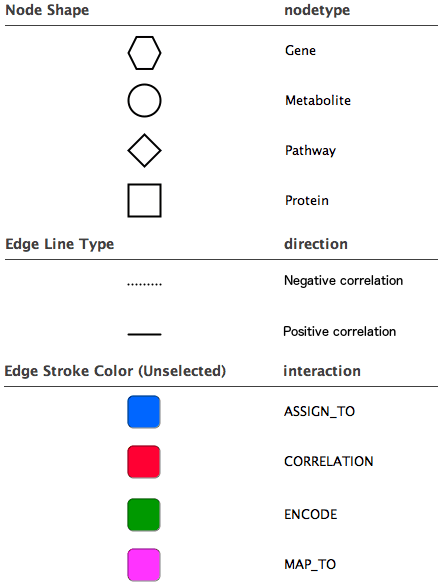
Diagram legend
References
Metabolomics and transcriptomics data used in this example are taken from the following publication:
- Wikoff WR, et al. Metabolomic markers of altered nucleotide metabolism in early stage adenocarcinoma. Cancer Prev Res (Phila) 2015;8(5):410-8.
Go to HOME | Documentation | fetchGrinnNetwork | fetchCorrGrinnNetwork | fetchDiffCorrGrinnNetwork | fetchModuGrinnNetwork | fetchGrinnCorrNetwork | input formats